

Kyazmaは、遺伝子連鎖解析に特化したソフトウェアです。Kyazmaのソフトウェア製品であるJoinMapは、二倍体実験集団における遺伝子連鎖マップの計算のためのMS-Windowsプログラムであり、MapQTLは、量的形質の連鎖解析のためのソフトウェアです。Kyazmaは、植物遺伝学において広く使用されており、多くの植物種の遺伝研究に利用されています。また、果樹や森林樹木などの多くの種類においても、遺伝子連鎖解析が強化されました。
【製品について】
■JoinMap
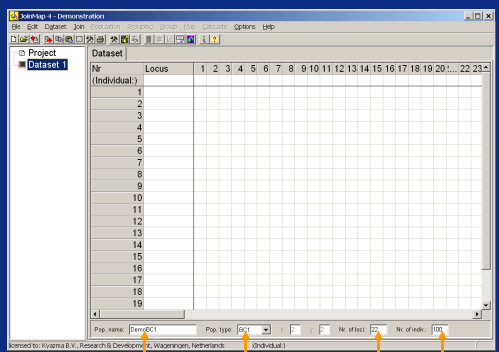
Kyazmaのソフトウェア製品であり、二倍体実験集団における遺伝子連鎖マップの計算のためのMS-Windowsプログラムです。JoinMapは、交雑種の2つの個体間の交配による完全同胞家族(F1)を含む一般的な実験集団に対応できます。JoinMapは、実験データの詳細な研究を可能にする高品質なツールを提供します。直感的なユーザーインターフェースは、データのより良い探索を促します。
■MapQTL
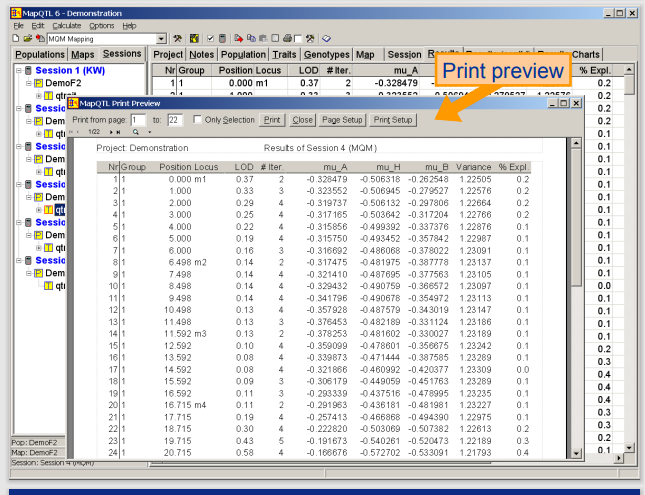
Kyazmaのソフトウェア製品であり、二倍体実験集団における量的形質遺伝子座(QTL)のマッピングのためのコンピュータプログラムです。MapQTLは、インターバルマッピングを用いたQTL実験を強力なMQMマッピング(コンポジットインターバルマッピング)もしくはノンパラメトリック法により解析することができます。MapQTLは、MS-Windowsプログラムであり、使いやすく、非常に高速であり、解析結果を表や(調整可能な)グラフで表示し、MS-Windowsテキスト処理およびプレゼンテーションソフトウェアにエクスポートすることができます。
【ライセンスについて】
マイナーチェンジは無料でVerup可能ですが、メジャーチェンジの際は、Upgradeが必要です。
メーカーの製品サイト
https://www.kyazma.nl/
【種別】製品
【言語】英語
【動作環境】Windows
【製品について】
■JoinMap
Kyazmaのソフトウェア製品であり、二倍体実験集団における遺伝子連鎖マップの計算のためのMS-Windowsプログラムです。JoinMapは、交雑種の2つの個体間の交配による完全同胞家族(F1)を含む一般的な実験集団に対応できます。JoinMapは、実験データの詳細な研究を可能にする高品質なツールを提供します。直感的なユーザーインターフェースは、データのより良い探索を促します。
■MapQTL
Kyazmaのソフトウェア製品であり、二倍体実験集団における量的形質遺伝子座(QTL)のマッピングのためのコンピュータプログラムです。MapQTLは、インターバルマッピングを用いたQTL実験を強力なMQMマッピング(コンポジットインターバルマッピング)もしくはノンパラメトリック法により解析することができます。MapQTLは、MS-Windowsプログラムであり、使いやすく、非常に高速であり、解析結果を表や(調整可能な)グラフで表示し、MS-Windowsテキスト処理およびプレゼンテーションソフトウェアにエクスポートすることができます。
【ライセンスについて】
マイナーチェンジは無料でVerup可能ですが、メジャーチェンジの際は、Upgradeが必要です。
メーカーの製品サイト
https://www.kyazma.nl/
【種別】製品
【言語】英語
【動作環境】Windows


